Contributed by Corinne Liying Lee, Ramamoorthy Rajkumar, Gavin Stewart Dawe, Department of Pharmacology, Yong Loo Lin School of Medicine, National University Health System; Neurobiology and Ageing Programme, Life Sciences Institute, National University of Singapore; and Singapore Institute for Neurotechnology (SINAPSE), Singapore 117456
The nucleus incertus (NI) is a distinct group of cells in the pontine periventricular gray, adjacent to the dorsal tegmental nucleus.[1-3] Interest in this small and distinct group of neurons spiked after it was shown to express high levels of corticotropin releasing hormone receptor 1 (CRF1),[4-5] which suggests a role for the NI in the circuitry of stress responses. While the NI also expresses neuromedin B [6-7] and cholecystokinin,[3,8] expression of relaxin-3 overlaps that of CRF1 and is highly specific and mostly concentrated in the NI. As such, the NI is considered the principle source of relaxin-3 in the mammalian brain and possibly serves as a key point of regulation of the relaxin-3 neural circuitry. The exact function of the NI and relaxin-3 is still largely unknown but the NI has been implicated in the control of multiple neural networks, ranging from the regulation of feeding behavior,[9-11] to the modulation of stress and anxiety [2,12-13] and cognition.[14-16] In our recent study published in Brain Research, we demonstrated a method for the selective ablation of CRF1-expressing neurons in the NI using the CRF-SAP conjugate (CRF-SAP; Cat. #IT-13).[17]
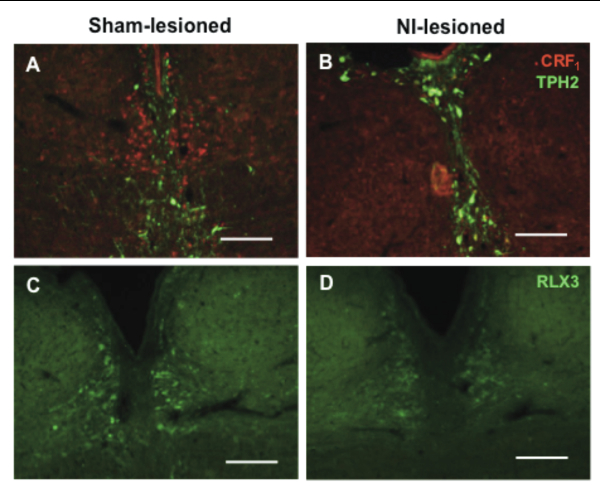
While some of the hypothesized functions of the NI are speculations from anatomical mapping of its vast connections to the rest of the brain, most of the experimental evidence to date stems from studies using relaxin-3 related peptides, studies of neural activation based on behavioral activity, electrical stimulation or infusion of CRF. To study the role of the NI, bilateral injections of 86 ng/site of CRF-SAP were made into the NI to selectively ablate the CRF1-expressing neurons in the NI. As a control, sham lesions were made by infusion of Blank-SAP (Cat #IT-21). Verification of the lesion after 14 days of recovery, revealed a significant and specific reduction in CRF1 and relaxin-3 positive cells in the NI (See Cover Figure). These NI-lesioned rats also exhibited a significantly greater freezing period when compared to sham-lesioned rats when tested in a cued fear conditioning paradigm.
Our results suggest that CRF-SAP selectively targets and kills CRF1-expressing cells in the NI, consistently decreasing relaxin-3 levels in the NI and one of its known projection targets, the medial septum (MS). Taken together, these data indicate that this CRF1 receptor-targeted lesion model is a valuable tool for studying the relaxin-3 circuitry in the brain. The clear behavioral deficit observed also supports the use of CRF-SAP to selectively perturb the NI and thus provides an additional approach to study the NI and the relaxin-3 system in behavioral neuroscience. The approach to selectively lesion relaxin-3 positive cells in the NI using CRF-SAP expands the spectrum of use of targeted toxins to study the function of discrete neuron subgroups in other brain structures.
References: (back to top)
- Goto M, Swanson LW, Canteras NS. J Comp Neurol 438(1):86-122, 2001.
- Burazin TC, Bathgate RA, Macris M, Layfield S, Gundlach AL, Tregear GW. J Neurochem 82(6):1553–1557, 2002.
- Olucha-Bordonau FE, Teruel V, Barcia-Gonzalez J, Ruiz-Torner A, Valverde-Navarro AA, Martinez-Soriano F, J Comp Neurol 464(1):62–97, 2003.
- Potter E, Sutton S, Donaldson C, Chen R, Perrin M, Lewis K, Vale W. Proc Natl Acad Sci USA 91(19):8777–8781, 1994.
- Bittencourt JC, Sawchenko PE. J Neurosci 20(3),:1142–1156, 2000.
- Chronwall BM, Skirboll LR, O’Donohue TL. Neurosci Lett 53(1):109–114, 1985.
- Wada E, Way J, Lebacq-Verheyden AM, Battey JF. Eur J Neurosci 38(4):2516–2525, 1990.
- Kubota Y, Inagaki S, Shiosaka S, Cho HJ, Tateishi K, Hashimura E, Tohyama M. Neuroscience 9(3):587–604, 1983.
- McGowan BM, Stanley SA, Smith KL, White NE, Connolly MM, Thompson EL, Bloom SR. Endocrinology 146(8):3295–3300, 2005.
- McGowan BM, Stanley SA, Smith KL, Minnion JS, Donovan J, Thompson EL, Bloom SR. Regul Pept 136(1-3):72–77, 2006.
- Rajkumar R, See LK, Dawe GS. Brain Res 1508:34–43, 2013.
- Smith CM, Hosken IT, Sutton SW, Lawrence AJ, Gundlach AL. Genes Brain Behav 11(1):94–104, 2012.
- Watanabe Y, Tsujimura A, Takao K, Nishi K, Ito Y, Yasuhara Y, Tanaka M. Behav Neurosci 5:50.6, 2011.
- Ma S, Olucha-Bordonau FE, Hossain MA, Lin F, Kuei C, Liu C, Gundlach AL. Learn Mem 6(11):730–742, 2009.
- Cervera-Ferri A, Guerrero-Martinez J, Bataller-Mompeán M, Taberner-Cortes A, Martinez-Ricós J, Ruiz-Torner A, Teruel-Marti V. Exp Brain Res 211(2):177-192, 2011.
- Farooq U, Rajkumar R, Sukumaran S, Wu Y, tan WH, Dawe GS. Eur J Neurosci 38(4):2516-2525, 2013.
- Lee LC, Rajkumar R, Dawe GS. Brain Res 1543:179-190, 2014.