Contributed by Dr. Colm Cunningham, Trinity College, Institute of Neuroscience & School of Biochemistry and Immunology, Dublin, Ireland
It is well established that peripheral inflammation can signal the intact CNS to bring about adaptive changes in behavior during the sickness response. However, during aging and dementia, the brain is particularly susceptible to the deleterious effects of such insults. Delirium is an acute and severe disturbance in cognitive function with particular deficits in attention, memory, orientation and perception. The pathophysiology of delirium remains poorly understood but it is clear that dementia and prior cognitive impairment are major risk factors for delirium, and systemic inflammation is a frequent trigger. Hypocholinergia and increased neuroinflammation have been proposed as major players in delirium pathophysiology but no clinical or animal studies have investigated the interplay between systemic inflammation and cholinergic function.
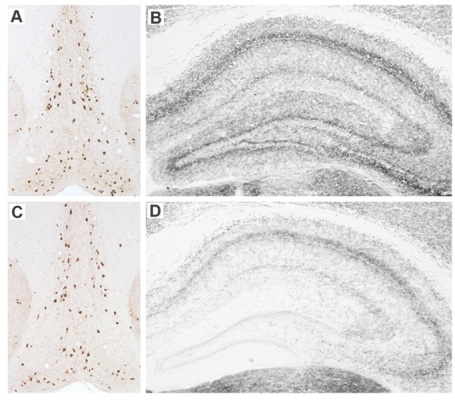
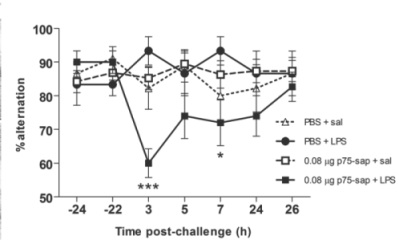
In a paper published in The Journal of Neuroscience,[1] we hypothesized that making limited lesions of the basal forebrain cholinergic neurons (BFCN) would leave these animals vulnerable to the CNS effects of systemic inflammation. The BFCN, which comprises the medial septum, the diagonal band and the nucleus basalis, is the source of most acetylcholine in the forebrain and this area degenerates markedly during Alzheimer’s disease. To specifically lesion this region we used the ribosomal toxin saporin, covalently linked to an antibody directed against the p75 neurotrophin receptor that is highly expressed on BFCN. We showed that intracerebroventri-cular (icv) injection of this toxin (mu p75-SAP, Cat. #IT-16) at low doses could induce approximately 20% destruction of the cholinergic cells of the medial septum without obvious effect on working memory function (see Fig 1). However, if these animals were allowed to recover for 40 days post-surgery, and then challenged systemically with bacterial endotoxin to mimic mild-moderate systemic bacterial infection (100 µg/kg LPS intraperitoneal), only those animals with prior lesions showed acute working memory deficits.
Many of our previous predictions about the interaction of prior pathology and systemic inflammation [2] were based on the original demonstration of microglial priming and subsequent exaggerated inflammatory responses to systemic inflammatory insult.[3] However, despite clear microglial activation at three days after mu p75-SAP lesions, microglial activation had resolved by 40 days post-lesion, and at this time microglia also did not show exaggerated inflammatory cytokine induction after systemic inflammation (Fig 2). Thus, the acute working memory deficits appear to occur in the absence of microglial priming. This implies that the ‘vulnerability’ in these animals represents a neuronal susceptibility to disruption of function. We showed, using the acetylcholine muscarinic receptor antagonist scopolamine, that the T-maze working memory task used in these studies was indeed dependent on cholinergic function and further showed that treatment with the acetylcholinesterase inhibitor donepezil, one hour after LPS, protected against the working memory deficit observed. Collectively these data show that the loss of 20% of cholinergic neurons of the basal forebrain cholinergic system does not robustly affect cognitive function under normal conditions, but leaves these animals vulnerable to significant cognitive disruption upon an acute systemic inflammatory insult.
We believe that these findings have significant implications for delirium, particularly in the setting of existing cognitive impairment/dementia and that this limited cholinergic lesion model will be an extremely useful tool in delineating molecular pathways to dysfunction arising from systemic inflammatory insults in older or cognitively-impaired individuals.
References: (back to top)
- Robert H. Field, Anna Gossen, and Colm Cunningham (2012) Prior Pathology in the Basal Forebrain Cholinergic System Predisposes to Inflammation-Induced Working Memory Deficits: Reconciling Inflammatory and Cholinergic Hypotheses of Delirium. J Neurosci 32(18):6288–6294.
- Murray C, Sanderson DJ, Barkus C, Deacon RM, Rawlins JN, Bannerman DM, Cunningham C (2012) Systemic inflammation induces acute working memory deficits in the primed brain: relevance for delirium. Neurobiol Aging 33:603– 616.e3.
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH (2005) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25:9275–9284.