Contributed by Maria Montero (a), Berta Gonzalez (b), Jens Zimmer (a)
(a) Anatomy and Neurobiology, University of Southern Denmark, Denmark
(b) Histology Unit, Autonomous University of Barcelona, Spain
Microglial cells contribute about 12% of the cells in the brain, acting as “biosensors” for homeostatic regulation in normal and pathological conditions.[3,13] Resting microglial cells in the adult brain have thin ramified processes, but transform into a non-phagocytic activated phenotype, or a phagocytic activated phenotype when activated.[14] Upon activation, structural changes towards an amoeboid appearance occur with transformation of the normal fine processes into short coarser processes [4,14] in parallel with upregulation of normal markers like Mac-1,[12] MHC class I and II molecules [5] and secretion of cytokines like interleukin-1beta (IL-1ß), [2] transforming growth factor beta-1 (TGF-ß1) [6] and tumor necrosis factor alpha (TNF-α).[1] The reactions to injury also include a drastic increase in the number of microglial cells at the lesion site, due to migration and local proliferation.[10] However, it is still not clear to what extent or under which conditions activated forms of microglia exert positive or negative effects on neuronal survival.[7] Experimental depletion of microglial cells, before application of a standardized neurodegenerative insult, would, however, help clarify the protective or degenerative role of microglial cells.
Depletion of microglial cells in mouse brain hippocampal slice cultures was successfully achieved by adding a microglia-targeting toxin, Mac-1-SAP (a conjugate of saporin and a CD11b antibody, Cat. #IT-06) to the culture medium for 7 days (Fig. 1).[8]
For experimental testing of the effects of microglia depletion on ischemia-induced neurodegeneration, Mac-1-SAP treated and corresponding untreated hippocampal slice cultures were subjected to standardized 30-min oxygen-glucose deprivation (OGD), mimicking transient cerebral ischemia and preferentially causing the more susceptible CA1 pyramidal cells to degenerate. The resulting neuronal cell death was quantified by densitometric measurements of the cellular uptake of the fluorescent intercalating agent propidium iodide (PI) in the CA1 area [11] (Fig. 2). Other Mac-1-SAP treated and control cultures were stained immunohistochemically for neuronal, astroglial and microglial markers 1, 7 or 14 days after OGD (Fig. 3).
Twenty-four hours after OGD, the Mac-1-SAP treated, microglia-depleted hippocampal slice cultures displayed, in comparison to correspondingly OGD-exposed control cultures, a significant increase in CA1 pyramidal cell death, as monitored by cellular PI uptake (Fig. 2), as well as increased astroglial reactivity. At the same time point, the OGD lesioning induced an increase in Mac-1-positive cells in the microglia-depleted cultures, meaning that the few visible Mac-1 cells remaining after the immunotoxic treatment (and possible additional ones with staining below detection level) did, as normally, respond to the ischemia-like insult by stronger staining for microglial markers (Fig. 3). Using BrdU-labeling of dividing cells we also demonstrated lesion-induced proliferation of the few remaining microglial cells. [8] At 7 and 14 days after OGD the differences between Mac-1-SAP treated and non-treated cultures remained noticeable in terms of more neuron loss in cultures deprived of microglia, while the astroglial reactivity seemed to even out. During the same period more microglial cells appeared in the originally deprived cultures at the same time as the appearance of the cells normalized (Fig. 3 G, H). Such reoccurrence over time has also been noted for depleted cultures left to survive for an additional 2-3 weeks.
Conclusions: (1) Exposure of mouse hippocampal slice cultures to Mac-1-SAP efficiently eliminates almost all microglial cells present in the cultures. (2) Mac-1-SAP treatment does not by itself induce increased neuronal cell death as monitored by cellular uptake of PI. (3) When subjected to an ischemia-like transient oxygen-glucose deprivation (OGD), Mac-1-SAP treated cultures displayed an increase in CA1 pyramidal cell death, as compared to non- treated OGD-lesioned cultures.
Perspectives: Brain slice cultures are easily accessible for microscopical inspection and addition of compounds and, as such, widely used as experimental models for CNS injury and disease and for screening of neurotoxic and neurotrophic compounds. Efficient and controlled depletion of microglial cells by Mac-1-SAP in slice cultures is a valuable tool for studies of microglial interactions with other cell types in the CNS, and microglia-mediated actions of, for example, anti-inflammatory compounds. [8]
Acknowledgments: Figures were published in Brain Research, Montero M., González B., Zimmer J. Immunotoxic depletion of microglia in mouse hippocampal slice cultures enhances ischemia-like neurodegeneration, in press. Copyright Elsevier, 2009.
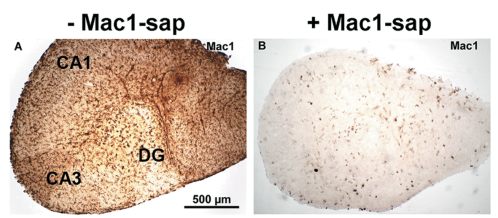
Scale bar valid for both A and B.


Figure 2. Left panel: Fluorescent micrographs of distribution and density of cellular uptake of PI in control (A, C) and OGD-lesioned mouse hippocampal slice cultures (B, D) with normal microglial content (A, B) and prelesional depletion of microglia (C, D). The OGD-induced cell death primarily affects CA1 pyramidal cells (CA1).
Right panel: Densitometric measurements of the cellular uptake of PI in CA1 pyramidal layer of untreated and Mac-1-SAP treated slice cultures as well as corresponding cultures 24 hours after 30 min. of OGD. Data are shown as a percentage of PI uptake standardized to OGD (n = 11-18) (*** p < 0.001).

Figure 3. Microglial depletion and reactions to 30 min OGD in CA1 area of mouse hippocampal slice cultures, visualized by Mac-1 immunohistochemistry. Treatment of 1 week old slice cultures with the Mac- 1-SAP complex for 1 week dramatically reduced the number of microglial cells (A and E). One day after OGD, the number of reactive, Mac1-positive microglial cells increased in both normal (non-depleted) (B) and depleted cultures (F), followed by a normalization in morphology and staining density over the next 2 weeks, which also included the originally depleted slice cultures (E, F, G, H).
References: (back to top)
- Buttini M, Appel K, Sauter A, Gebicke-Haerter PJ, Boddeke HW (1996) Neuroscience 71:1-16.
- Buttini M, Sauter A, Boddeke HW (1994) Brain Res Mol Brain Res 23:126- 134.
- Dheen ST, Kaur C, Ling EA (2007) Curr Med Chem 14:1189-1197.
- Jensen MB, Finsen B, Zimmer J (1997) Exp Neurol 143:103-116.
- Kato H, Kogure K, Araki T, Itoyama Y (1995) Brain Res 694:85-93.
- Lehrmann E, Kiefer R, Christensen T, Toyka KV, Zimmer J, Diemer NH, Hartung HP, Finsen B (1998) Glia 24:437-448.
- Li L, Lu J, Tay SS, Moochhala SM, He BP (2007) Brain Res 1159:8-17.
- Montero M, Gonzalez B, Zimmer J (2009) Experimental Neurology 218(1):11-23.
- Montero M, González B, Zimmer J (2009) Brain Research, in press.
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K (2006) Faseb J 20:714-716.
- Noraberg J, Poulsen FR, Blaabjerg M, Kristensen BW, Bonde C, Montero M, Meyer M, Gramsbergen JB, Zimmer J (2005) Curr Drug Targets CNS Neurol Disord 4:435-452.
- Perry VH, Hume DA, Gordon S (1985) Neuroscience 15:313-326.
- Schloendorn J, Sethe S, Stolzing A (2007) Rejuvenation Res 10:87-99.
- Streit WJ, Walter SA, Pennell NA (1999) Prog Neurobiol 57:563-581