Contributed by Dr. Thomas Beach, Sun Health Research Institute, Sun City, AZ
Our hypothesis, developed through our own human and animal studies1-6 and the cell culture work of others (beginning with Nitsch7), is that the normal, age-related loss of cortical cholinergic innervation leads to Aβ deposition and Alzheimer’s disease. To test this hypothesis, we have been using a saporin- conjugated antibody to lesion the cholinergic basal forebrain (CBF) of rabbits. The antibody is the ME20.4 monoclonal8 against the low affinity nerve growth factor receptor, also known as the p75 neurotrophin receptor (p75NTR). This approach had already been shown to result in an effective and specific lesion in rats, but we wished to use an animal with an Aβ sequence identical to that of humans (the rat sequence differs by 3 amino acids), since we considered that, because of this difference, rats may be less likely to produce Aβ deposits. As the IgG192 anti-p75NTR antibody used for rats does not recognize rabbit CBF neurons, we tested the ME20.4 antibody and found, in preliminary immunohistochemical studies, that it stained the rabbit CBF neurons beautifully (Fig 1c,d). We obtained the ME20.4-producing cells from the American Type Culture Collection (ATCC) and forwarded these to Advanced Targeting Systems for antibody production and coupling to saporin.
We injected 30-40 μl of the conjugate into the lateral ventricles of New Zealand white rabbits and sacrificed the animals at 5 weeks, 3 months and 6 months. At all time intervals, we found that lesioned animals showed loss of CBF neurons (Fig 1a-d), cortical ChAT decrements of 50-70%, and profound loss of cholinergic nerve fibers (Fig 1e,f). All lesioned animals showed Aβ deposition in cerebral blood vessel walls and as perivascular diffuse deposits in the neuropil (Figure 1g,h). Biochemical (ELISA) measurements confirmed that Aβ concentrations were elevated in the cerebral cortex, 2.5-fold and 8-fold, respectively, for the forms of the peptide ending in 40 and 42. This model may recapitulate the events leading to Aβ deposition in aging and the sporadic form of Alzheimer’s disease.
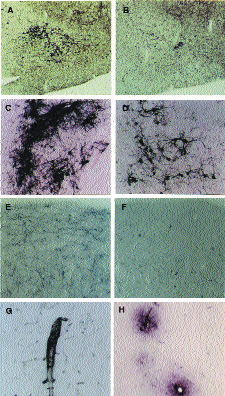
A,B: rabbit nbm area in control (A) and immunotoxin- treated animals (B), stained for AChE. Note the marked depletion of nbm neurons in the latter.
C,D: rabbit nbm in control (C) and treated (D) animal, stained immunohistochemically for p75 NTR. Note the depletion of neurons in the treated animal.
E,F: frontal cortex from control (E) and treated (F) animals, stained for AChE. Note eradication of cholinergic fibers in the latter.
G,H: Aβ deposition in cerebral cortex of lesioned animals. Aβ is deposited in blood vessel walls (G) and in the perivascular neuropil (H).
References
- Beach TG and McGeer EG. Acta Neuropathol. 83:292-299, 1992.
- Beach TG, Walker, D.G., Cynader MS and Hughes LH. Neurosci. Lett. 142:253-256, 1996.
- Beach TG, Hughes LH and Honer, WG. Acta Neuropathol. 93:146-153, 1997.
- Beach, TG, Sue, LI, Scott, S and Sparks, DL. Alz. Reports 1:375-380, 1998.
- Beach, TG, Potter, PE, Kuo, Y-M, Emmerling, MR, Durham, RA, Webster, SD, Walker, DG, Sue, LI, Scott, S, Layne, KJ and Roher, AE. Neurosci. Lett. 283: 9-12, 2000.
- Beach, TG, Kuo, Y-M, Spiegel, K, Emmerling, MR, Sue, LI, Kokjohn, K and Roher, AE. J. Neuropathol. Exp. Neurol. 59:308-313, 2000.
- Nitsch, RM, Slack, BE, Wurtman, RJ, and Growdon, JH. Science 258:304-307, 1992.
- Fine, A, Hoyle, C, Maclean, CJ, Levatte, TL, Baker, HF and Ridley, RM. Neuroscience 81:331-343, 1997.
Product information related to cover article: ME20.4-SAP, Cat. #IT-15