Contributed by Francesco Ferrini (1) and Yves De Koninck (2,3)
1) Department of Veterinary Sciences, University of Turin,10095 Grugliasco, Turin, Italy
2) Institut universitaire en santé mentale de Québec, QC, G1J 2G3, Canada
3) Department of Psychiatry and Neuroscience, Université Laval, Québec, QC, G13 7P4, Canada
Morphine-induced hyperalgesia and tolerance dramatically limit the use of morphine, especially in chronic diseases. By definition, morphine tolerance is a reduced antinociceptive effect for a given morphine dose, while morphine-induced hyperalgesia is a state of nociceptive sensitization observed in morphine-treated patients.[1,2] It is therefore tempting to postulate that antinociceptive tolerance is set by the decrease in nociceptive threshold due to the hyperalgesia.[3] However, this common view appears to be in contrast with clinical evidence indicating that while increasing the morphine dose can effectively counteract morphine tolerance, the same approach can backfire and worsen pain symptoms in patients with morphine-induced hyperalgesia.[1] In our recent study published in Nature Neuroscience,[4] we demonstrated that morphine hyperalgesia and morphine tolerance are mechanistically distinct and that morphine induces hyperalgesia by recapitulating the same maladaptive mechanisms in the spinal cord observed in pathological pain syndromes.
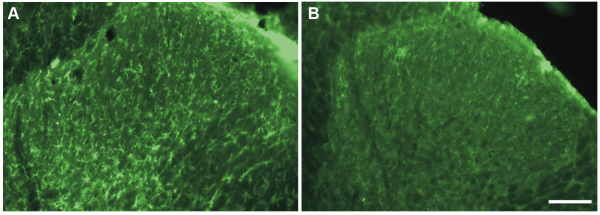
In particular, we addressed the question whether microglia drive morphine-induced hyperalgesia, as the communication between neurons and microglia in the spinal dorsal horn plays a central role in the development of neuropathic pain.[5] The role of microglia in diseases can be tested by using pharmacological tools, such as minocycline, which have been proved to inhibit microglia function.[6] However, the specificity of such approaches to target microglia is debated and direct effects on neuronal activity cannot be ruled out.[7] Therefore, we decided to perform intrathecal injections of a saporin-conjugated antibody against the macrophage-1 antigen (Mac-1, 16–32 μg; Mac-1-SAP rat, Cat #IT-33) to selectively ablate spinal microglia in rats with established morphine-induced hyperalgesia. Injections of Mac-1-SAP or saporin alone as control (SAP, 20 μg; Cat #PR-01) were initiated after seven days of morphine treatment and performed at lumbar level once a day for three days. Mac-1-SAP significantly reduced the level of CD11b expression in the lumbar spinal dorsal horn (see Fig.1) and the treatment reversed mechanical and thermal hypersensitivity induced by morphine. Conversely, the immunotoxin did not affect the development of morphine tolerance in the same animals.
These findings point out the specific role of microglia in the development of pain hypersensitivity following morphine treatment. Thereafter, we dissected the underlying molecular mechanisms and found that morphine induced P2X4 receptor upregulation in spinal microglia, which in turn triggered the synthesis and release of brain-derived neurotrophic factor (BDNF). Microglial BDNF has been shown to induce pain hypersensitivity in spinal neurons by hampering the function of the K+-Cl– co-transporter KCC2, the main neuronal chloride extruding transporter in neurons, via trkB signaling.5 Consistently, we found that such BDNF-trkB-KCC2 signaling cascade is activated by morphine and alters chloride-mediated inhibition on spinal neurons, thus increasing network excitability.
All together, our study indicates that morphine-induced hyperalgesia, as neuropathic pain, is a pathological alteration of pain sensitivity whose expression is gated by spinal microglia. Targeting at any level, this microglia-to-neuron cascade is a valuable strategy to improve the use of morphine in chronic pain.
References: (back to top)
- Lee M., Silverman SM, Hansen H, Patel VB, Manchikanti, L. (2011) A comprehensive review of opioid-induced hyperalgesia. Pain Physician 14:145–161.
- Crofford LJ. (2010) Adverse effects of chronic opioid therapy for chronic musculoskeletal pain. Nat Rev Rheumatol 6:191-7.
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP Jr, Lai J, Porreca F. (2001) Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci 21:279–286.
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del’Guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De Koninck Y. (2013) Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl- homeostasis. Nat Neurosci 16:183-92.
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438:1017-1021.
- Yoon SY, Patel D, Dougherty PM. (2012) Minocycline blocks lipopolysaccharide induced hyperalgesia by suppression of microglia but not astrocytes. Neuroscience 221:214-24.
- González JC, Egea J, Del Carmen Godino M, Fernandez-Gomez FJ, Sánchez-Prieto J, Gandía L, García AG, Jordán J, Hernández-Guijo JM. (2007) Neuroprotectant minocycline depresses glutamatergic neurotransmission and Ca(2+) signalling in hippocampal neurons. Eur J Neurosci 26:2481-95.