Contributed by Ekaterina Likhtik Columbia University,
New York Psychiatric Institute, 1051 Riverside Drive, New York, NY 10032
Introduction by Douglas Lappi, Ph.D., President/CSO: This quarter’s Cover Article is from Ekaterina Likhtik who reprises her recently published work in Nature 454:642-645. In the News and Views, Sah and Westbrook comment that “Neuronal receptors in these circuits—such as those targeted with saporin in Likhtik and colleagues’ study—are likely to become targets for the development of specific treatments for many anxiety disorders.”
The amygdala is a key subcortical structure in the neural circuit that processes acquisition as well as extinction of conditioned fear. Although a large body of literature details how amygdala activity results in fear conditioning, fear extinction circuits are less well understood. In particular, it is difficult to study the role that one of its potentially important cell groups, the intercalated (ITC) cells, may play in this behavior. These cells likely constitute an important interface between the input and output nuclei of the amygdala, gating information flow out of the amygdala during fear conditioning and extinction.[1,2]
To date the obstacle to studying the ITC cells is that they occur in small, anatomically distributed clusters [3] and are therefore difficult to selectively lesion using conventional methods. In order to circumvent this issue, we took advantage of the high levels of µ-opioid receptor expression observed in ITC cells in the light microscope. Indeed, a more detailed analysis using electron microscopy, revealed that the ITC cells express µ-opioid receptors post-synaptically at 3-6 times the rate of surrounding amygdala nuclei (Fig 1b). Given this imbalance in receptor expression, we were confident that local injections of the µ-opioid receptor-targeted Dermorphin-SAP (Cat. #IT-12) would be the optimal way of testing the impact of these cells on behavior, without compromising the integrity of the surrounding tissue.
To test whether the ITC cells play a role in extinction expression, rats were habituated and fear conditioned in one context, and then extinguished in a second context. One day after extinction, animals underwent surgery where they received bilateral infusions stereotaxically aimed at the ITC cells of either Dermorphin-SAP (3 pmol/µl, infusion rate .01 µl/min, total of 0.25 µl per hemisphere) or an equivalent amount of control–a non-targeted peptide conjugated to saporin, Blank-SAP (Cat. #IT-21; Fig 1a). This protocol did not result in any adverse effect on post-operative weight gain, posture or exploratory behavior.
After recovery, rats were tested for how well they remembered extinction training. During the extinction recall session, sham-lesioned animals displayed low freezing to presentations of the conditioned stimulus, consistent with having retained the extinction training. On the other hand, animals with ITC cell lesions had a dramatic increase in the amount of freezing to the previously extinguished conditioned stimulus, indicating that their recall of the previous session was substantially diminished (Fig 2a). Importantly, rats that received identical Dermorphin-SAP infusions in nuclei surrounding the ITC cells, recalled extinction training as well as controls, suggesting an ITC-specific effect.
Stereological cell counts revealed that properly placed tracks delivering Dermorphin-SAP decreased the number of ITC cells in lesioned animals by 34% as compared to sham-lesioned controls (Fig 2b). In contrast, cells in the adjacent central nucleus of the amygdala were unaffected by the lesion. In addition, an inverse correlation between the number of surviving ITC cells and freezing during extinction recall (r = – 0.67) was observed, whereas there was no such correlation between the number of cells in the central nucleus and freezing during extinction recall (r = – 0.13).
In this study, Dermorphin-SAP has served as an important tool to seek out a potentially useful target for clinical intervention. Dermorphin-SAP lesions have allowed us to safely and selectively eliminate a proportion of amygdala ITC cells, revealing the importance of these neurons in the expression of extinction. Given that extinction failure is a robust model for a number of anxiety disorders, [4] we can now explore ways of pharmacologically manipulating µ-opioid and other receptors expressed on the ITC cells to control amygdala output and facilitate extinction.
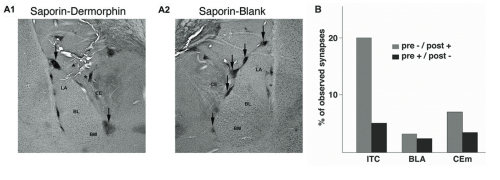
(A) Coronal sections from rats that received either Dermorphin-SAP (A1) or Blank-SAP (A2) injections in the vicinity of ITC cells. Arrows indicate the remaining ITC cell clusters in the two animals. Asterisks point to injection tracks. Note that only cell clusters adjacent to tracks are affected by the infusion.
(B) Electron microscopy: proportion of synapses where µ-opioid receptor immunoreactivity was found in post-synaptic (grey) or pre-synaptic (black) elements of the basolateral (BLA), medial central (CEm) or ITC nuclei.
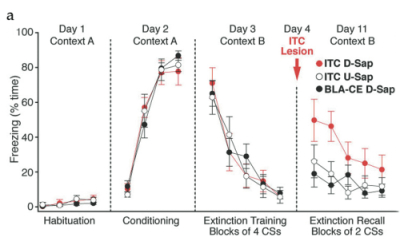
(A) Percent time freezing over experimental sessions in animals receiving Dermorphin-SAP injections in the ITC cells (red circles), in the BLA-CEA (black circles), or Blank-SAP injections in the ITC cells (white circles).
(B) Unbiased stereological estimates of cell numbers (mean ±sem, filled black circles) in the medial ITC cell clusters and central nucleus (CEA) in experimental (red circles) versus control (white circles) animals.
References: (back to top)
- Paré D, Quirk GJ, Ledoux JE. (2004) J Neurophysiol 92:1-9.
- Quirk GJ, Likhtik E, Pelletier JG, Paré D. (2003) J Neurosci 23:8800-8807.
- Millhouse OE (1986) J Comp Neurol 247:246-271.
- Ressler KJ, Mayberg HS (2007) Nat Neurosci 10:1116-1124.