Contributed by Jason J. Radley, Ph.D., Laboratory of Neuronal Structure and Function
The Salk Institute for Biological Studies and Foundation for Medical Research, La Jolla, CA 92037, USA
The medial prefrontal cortex (mPFC) has been proposed to play a role in the inhibition of hypothalamo-pituitary-adrenal (HPA) responses to emotional stress via influences on neuroendocrine effector mechanisms housed in the paraventricular hypothalamic nucleus (PVH) [1,2]. The locus coeruleus (LC) is the principal noradrenergic cell group in the brain, and plays established roles in promoting behavioral adaptations to a variety of alerting stimuli, including stressful ones [3,4]. While the PVH receives a substantial catecholaminergic innervation, the bulk of this arises not from the LC, but from medullary cell groups [5], which are implicated in mediating HPA responses to physiologic, but not emotional, stressors [6]. This raises the possibility that LC’s influence on stress-induced HPA activation might be mediated indirectly, through its projections to limbic and forebrain regions implicated in HPA control. The mPFC is involved in the processing of convergent cognitive and emotionally relevant information, and the LC noradrenergic projections to this region have been proposed to play a critical role in the modulation of working memory and attention [7]. These operations are likely to be involved the mPFC’s capacity to evaluate the contextual relevance and emotional valence of potentially threatening stimuli in order to effect adaptive responses. Nonetheless, the involvement of the LC-to-mPFC pathway in HPA regulation has not been tested, and is problematic, since dorsal mPFC (mPFCd) lesions have been shown to enhance [1,2], while LC lesions attenuate [8], HPA activation in response to acute emotional stressors.
In a recently published study [9], we assessed the effects of selectively ablating noradrenergic inputs into the mPFC, employing the axonally transported catecholamine immunotoxin, saporin-conjugated antiserum to dopamine-beta-hydroxylase (Anti-DBH-SAP, Cat. #IT-03), on acute restraint stress-induced activation of HPA output. Rats received injections of immunotoxin in the dorsal mPFC (centered in the prelimbic cortex), or sham injections of IgG-saporin or saline. 14 days thereafter, rats were subjected to 30 min of restraint stress and perfused 2 h later.
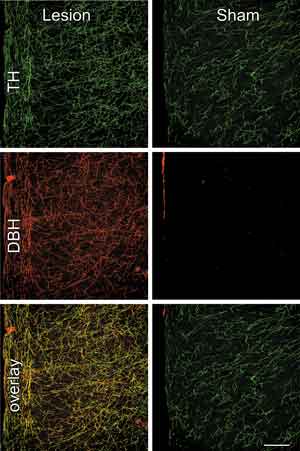
Anti-DBH-saporin injections virtually eliminated noradrenergic fibers and varicosities from the mPFCd, whereas control injections of the untargeted toxin (IgG-saporin, Cat. #IT-18) or aCSF left these inputs intact (Fig. 1, top). The specificity of the noradrenergic denervation of the mPFCd was assessed by examining the extent to which damage from the immunotoxin injection involved dopaminergic fibers and terminals in dual immunofluorescence preparations for tyrosine hydroxylase (TH) and DBH (Fig. 1). TH converts tyrosine to dihydroxyphenylalanine, a precursor of both dopamine (DA) and norepinephrine (NE). Immunolabeling of this enzyme represents both DA and NE fibers and terminals, whereas staining for DBH in cortex is specific to NE. Thus, the overlay of TH and DBH stained fibers and varicosities represents the subpopulation of inputs into mPFCd that are noradrenergic, whereas fibers singly-labeled for TH are dopaminergic (Fig. 1, middle). Following anti-DBH-saporin injections into PL, the density of dopaminergic (i.e., TH+/DBH-) fibers and varicosities was comparable to controls in density and distribution, while there was a near complete elimination of DBH staining in mPFCd, as well as of elements doubly-labeled for both enzymes (Fig. 1, bottom). Ancillary analyses revealed that immunotoxin injection in mPFCd resulted in a 23% decrease in the number of LC neurons and a corresponding decrease in stress-induced LC activational responses, compared to sham-lesioned controls (for more details, see [9]). These observations endorse anti-DBH-saporin as an effective tool for achieving focal noradrenergic denervation by ablating the neurons from LC that project to targeted terminal fields in mPFCd.
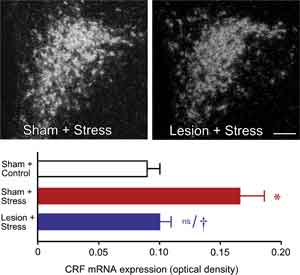
We initially surveyed the effects of immunotoxin lesions in mPFCd on stress-induced expression of Fos protein, a generic marker of neuronal activation, in the PVH. Acute stress resulted in a marked increase in Fos expression in the sham-lesioned animals, focused in the CRF-rich hypophysiotropic zone of PVH, and this effect was reduced by 28% in animals bearing immunotoxin lesions (P < 0.05). Ancillary analyses from sham- and immunotoxin-lesioned groups failed to reveal any effect of lesion status on the number of Fos immunoreactive neurons in the PVH of unstressed rats. To obtain an independent assessment of lesion effects on an endpoint more directly related to HPA activity, we examined relative levels of CRF mRNA in PVH using densitometry (Fig. 2, top). Consistent with the Fos expression data, restraint stress resulted in a twofold increase in CRF mRNA expression in the hypophysiotropic zone of PVH in sham-lesioned animals compared to unstressed controls (P < 0.001) (Fig. 2, bottom). In contrast, immunotoxin lesions diminished this effect (P < 0.005) to levels that did not differ significantly from those of unstressed controls (P < 0.007).
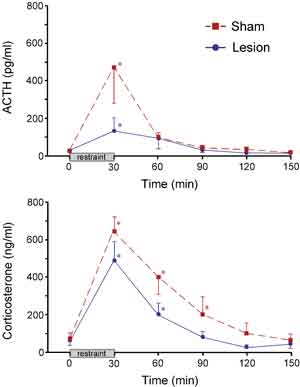
HPA secretory responses before and after the 30 min restraint stress were examined in separate groups of sham- and immunotoxin-lesioned animals (Fig. 3). Blood samples were obtained from indwelling jugular catheters that were implanted 2 days prior to stress exposure. Stress exposure significantly increased plasma levels of ACTH in both sham- and immunotoxin-lesioned animals (at 30 min). While these data suggest a difference between peak plasma levels of ACTH in immunotoxin- as compared to sham-lesioned animals, they did not differ significantly. Nonetheless, there was a significant reduction (by 62%; P < 0.05) in total integrated plasma ACTH levels in lesioned compared to sham groups, assessed by calculating areas under the curve. Stress exposure also significantly increased plasma levels of corticosterone in sham- and immunotoxin-lesioned animals. While there were no significant differences at any individual time point between sham and lesioned groups, there was an overall main effect for treatment (P < 0.05), and a decrease in integrated corticosterone levels (by 41%; P < 0.05) in lesioned as compared to unlesioned groups. Sham-lesioned animals also showed a prolonged increase in stress-induced plasma corticosterone levels (P < 0.05 at 30, 60, and 90 min compared with 0 min), whereas immunotoxin-lesioned animals show a more rapid recovery (P < 0.05 at 30 and 60 min).
The present findings localize previously documented HPA-facilitatory influences of LC, at least in part, to its projections to mPFCd and help to clarify the extended circuitry underlying mPFC modulation of HPA responses to acute emotional stress (Fig. 4). In addition to participating in the regulation of stress responses, the mPFC is also a target of them. Repeated exposure to emotional stress gives rise to dendritic atrophy and synapse loss in this region [10,11], findings that have clinical parallels in reports of mPFC shrinkage and functional impairment in posttraumatic stress disorder (PTSD) [12,13]. Norepinephrine has been linked to the mediation of maladaptive, as well as adaptive, consequences of stress exposure, being implicated in various psychiatric conditions, including PTSD [14]. Drugs that modulate noradrenergic transmission have demonstrated efficacy in treating such mood disorders via actions that may be exerted, at least in part, on the mPFC [15]. Further progress in unraveling the broader circuitry governing HPA responses to emotional stress, and the places of the LC and mPFC within it, should foster more informed management of stress-related psychiatric conditions.
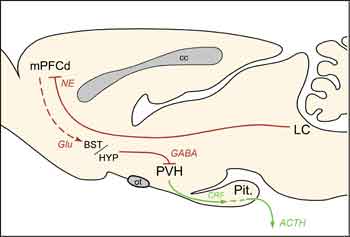
cc, corpus callosum; Glu, glutamate; Pit., pituitary gland; ot, optic tract.
References: (back to top)
- Diorio D, Viau V, Meaney MJ (1993) J Neurosci 13:3839-3847.
- Radley JJ, Arias CM, Sawchenko PE (2006) J Neurosci 26:12967-12976.
- Aston-Jones G, Rajkowski J, Cohen J (2000) Prog Brain Res 126:165-182.
- Sved AF, Cano G, Passerin AM, Rabin BS (2002) Physiol Behav 77:737-742.
- Sawchenko PE, Swanson LW (1982) Brain Res 257:275-325.
- Schiltz JC, Sawchenko PE (2007) J Comp Neurol 502:455-467.
- Aston-Jones G, Rajkowski J, Kubiak P, Valentino RJ, Shipley MT (1996) Prog Brain Res 107:379-402.
- Ziegler DR, Cass WA, Herman JP (1999) J Neuroendocrinol 11:361-369.
- Radley JJ, Williams BW, Sawchenko PE (2008) J Neurosci 28:5806-5816.
- Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, McEwen BS, Morrison JH (2004) Neuroscience 125:1-6.
- Radley JJ, Rocher AB, Rodriguez A, Ehlenberger DB, Dammann M, McEwen BS, Morrison JH, Wearne SL, Hof PR (2008) J Comp Neurol 507:1141-1150.
- Rauch SL, Shin LM, Segal E, Pitman RK, Carson MA, McMullin K, Whalen PJ, Makris N (2003) Neuroreport 14:913-916.
- Shin LM, Bush G, Whalen PJ, Handwerger K, Cannistraro PA, Wright CI, Martis B, Macklin ML, Lasko NB, Orr SP, Pitman RK, Rauch SL (2007) J Trauma Stress 20:701-712.
- Southwick SM, Bremner JD, Rasmusson A, Morgan CA, 3rd, Arnsten A, Charney DS (1999) Biol Psychiatry 46:1192-1204.
- Raskind MA, Peskind ER, Kanter ED, Petrie EC, Radant A, Thompson CE, Dobie DJ, Hoff D, Rein RJ, Straits-Troster K, Thomas RG, McFall MM (2003) Am J Psychiatry 160:371-373.
- Choi DC, Furay AR, Evanson NK, Ostrander MM, Ulrich-Lai YM, Herman JP (2007) J Neurosci 27:2025-2034.
- Roland BL, Sawchenko PE (1993) J Comp Neurol 332:123-143.