This article is a summary of data presented in reference #1. Figures 1-4 are taken from that article. This work was funded by NS045520 and DK40498 to S. Ritter.
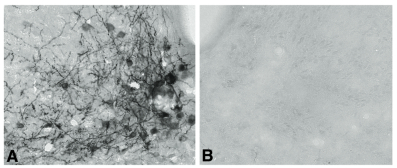
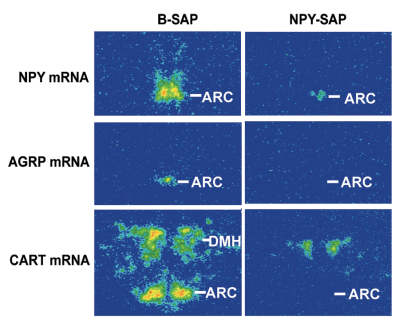
Neuropeptide Y (NPY) conjugated to saporin (SAP), a ribosomal toxin, is a compound designed to selectively target and lesion NPY receptor-expressing cells. We conducted competitive binding studies using I 125 -NPY to evaluate the binding of NPY-SAP (Cat. #IT-28) to rat forebrain homogenates (1). Results indicate that NPY-SAP binds to and has a higher binding affinity than NPY for the NPY receptor (Fig. 1). The binding results, in combination with previous studies demonstrating agonist-driven NPY receptor internalization (2, 3), indicated that this peptide-saporin conjugate would produce effective lesions of NPY receptor-expressing neurons. Accordingly, when we injected NPY-SAP (48 ng in 100 nl) bilaterally into the arcuate nucleus (ARC) of the hypothalamus, we found a profound reduction of NPY Y1 receptor-immunoreactivity (-ir) in the ARC (Fig. 2). We also found a nearly complete loss of NPY, AGRP and CART mRNA expression and α-MSH-ir in the ARC and mediobasal hypothalamus, showing that these NPY receptor-expressing neurons were lesioned by NPY-SAP (Fig. 3).
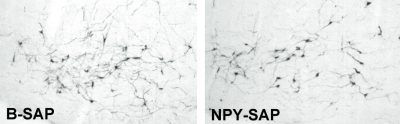
To date, there is no evidence that any of the available peptide-saporin conjugates are retrogradely transported. To determine whether NPY-SAP is retrogradely transported, we injected the conjugate into the ARC and examined catecholamine cell bodies in the A1/C1 region of the ventrolateral medulla. Nearly all of the catecholamine neurons in this area co-express NPY and project to the medial hypothalamus. A1/C1 neurons are almost completely destroyed by medial hypothalamic injections of the retrogradely transported immunotoxin, anti-dopamine-beta-hydroxylase-saporin (anti-DBH-SAP, Cat. #IT-03) (4-6). However, there was no loss of cells in this area in the NPY-SAP injected rats (Fig. 4), indicating that NPY-SAP is not internalized by NPY terminals or that, once internalized, there is no mechanism for retrograde transport of the conjugate. Supporting these findings, we also showed that NPY terminals in the area of cell body loss, though initially reduced, were not obliterated, as they would be if all NPY neurons innervating that area had been retrogradely destroyed.
In previous work, we used anti-DBH-SAP to examine the importance of hindbrain catecholamine neurons that innervated the hypothalamus for glucoregulation (7-10). We found that these neurons (many of which co-express NPY) are required for a number of glucoregulatory responses, including feeding, corticosterone secretion and suppression of estrous cycles in response to glucose deficit. In addition, we found that catecholamine neurons with projections to the spinal cord, which are distinct from those that project to the hypothalamus, are required for the adrenal medullary hyperglycemic response to glucoprivation. The goal of our work with NPY-SAP (1) was to determine whether the ARC NPY neurons, which co-express agouti-related protein (AGRP), are required for systemic glucoregulation. Gene knockout studies indicate that the NPY gene is required for glucoprivic feeding (11). However, there are multiple, presumably functionally heterogeneous, NPY populations in the brain. Furthermore, in the medial hypothalamus, the terminals of ARC NPY neurons are co-extensive with those of the hindbrain catecholamine neurons, making it difficult to distinguish the separate functions of these two NPY cell populations. NPY-SAP was useful in addressing this question. To date, we have examined feeding (Fig. 5), hyperglycemic and corticosterone responses to glucoprivation. None of these responses were impaired by ARC NPY-SAP injections that destroyed NPY receptor-expressing neurons, including the NPY/AGRP neurons, in the ARC and basomedial hypothalamus. However, these same lesions severely reduced feeding and body weight responses to leptin and feeding responses to ghrelin, which are known to depend upon ARC NPY receptor-expressing neurons. Thus, using anti-DBH-SAP and NPY-SAP we have been able to functionally differentiate the hindbrain NPY/catecholamine and the ARC NPY/AGRP co-expressing neuronal populations and to establish the primacy of the hindbrain NPY/catecholamine neurons for elicitation of systemic glucoregulatory responses.
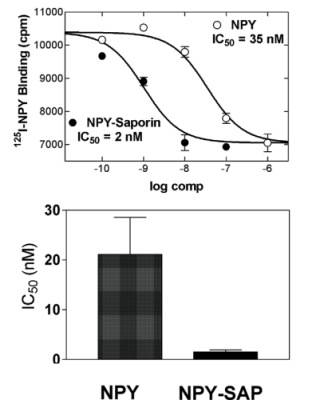
Top: Competitive binding of NPY and NPY-SAP with I125-NPY in rat forebrain tissue homogenates. Duplicate determinations were made for each concentration.
Bottom: Bars show IC50 for NPY and NPY reduced NPY-SAP binding. Data show that NPY-SAP has a binding affinity for NPY receptors that is equal to or greater than NPY at the concentrations examined.
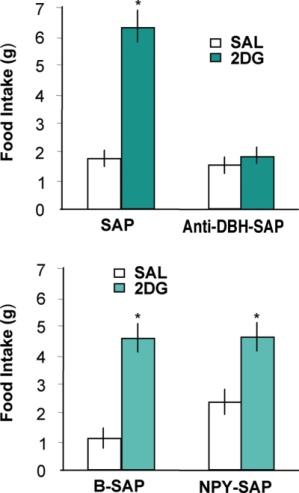
*2DG vs saline for the same group, P<.001
References: (back to top)
- Bugarith K, Dinh TT, Li AJ, Speth RC, Ritter S. (2005) Endocrinology 146:1179-1191.
- Parker MS, Parker SL, Kane JK. (2004) Regul Pept 118:67-74.
- Parker SL, Parker MS, Lundell I, Balasubramaniam A, Buschauer A, Kane JK, Yalcin A, Berglund MM. (2002) Regul Pept 107:49-62.
- Li AJ, Ritter S. (2004) Eur J Neurosci 19(8):2147-2154.
- Li AJ, Wang Q, Ritter S. (2006) Endocrinology 147(7):3428-3434.
- Ritter S, Bugarith K, Dinh TT. (2001) J Comp Neurol 432(2):197-216.
- Ritter S, Watts AG, Dinh TT, Sanchez-Watts G, Pedrow C. (2003) Endocrinology 144(4):1357-1367.
- Ritter S, Dinh TT, Sanders NM, Pedrow C. (2001) Soc Neuroscience Abstracts 27:947.3.
- I’Anson H, Sundling LA, Roland SM, Ritter S. (2003) Endocrinology 144(10):4325-4331.
- Hudson B, Ritter S. (2004) Physiol Behav 82(2-3):241-250.
- Sindelar DK, Ste Marie L, Miura GI, Palmiter RD, McMinn JE, Morton GJ, Schwartz MW. (2004) Endocrinology 145(7):3363-3368.